
Suppression of host oxidative burst is essential for survival of the intracellular parasite Leishmania donovani Screening of macrophage antioxidant enzymes during infection revealed marked upregulation of the heme-degrading enzyme, heme oxygenase-1 (HO-1). Moreover, HO-1-silenced RAW macrophages depicted increased superoxide production and decreased parasite survival. HO-1 induction decreased cellular heme content, thereby inhibiting the heme-dependent maturation of gp91phox, a catalytic component of major reactive oxygen species-producing enzyme NAD(P)H oxidase. Decreased gp91phox expression resulted in reduced stability of p22phox, another component of the catalytic center of NAD(P)H oxidase. Replenishing infected cells with exogenous heme reversed these effects and restored NAD(P)H oxidase activity. Persistent HO-1 expression at late hour of infection prompted us to investigate its effect on other host defense parameters, and inhibition study revealed a reciprocal relationship of HO-1 with host proinflammatory responses. Among all the HO-1-mediated heme degradation products (CO, Fe, and biliverdin), only CO documented potent anti-inflammatory effects. Quenching of CO during infection increased the production of disease-resolving cytokines IL-12 and TNF-α. Coimmunoprecipitation experiments revealed that CO inhibited the interaction of TLR4 with MyD88 and TIR domain-containing adapter-inducing IFN-β, thereby dampening the activation of NF-κB and IFN regulatory factor 3-mediated production of proinflammatory cytokines. Administration of HO-1 inhibitor tin protoporphyrin IX dichloride in infected BALB/c mice led to a decrease in liver and spleen parasite burden along with increased production of IL-12 and TNF-α. These results suggest that HO-1 on one hand inhibits reactive oxygen species generation and on the other hand downregulates host favorable cytokine responses, thereby facilitating intramacrophage parasite survival.
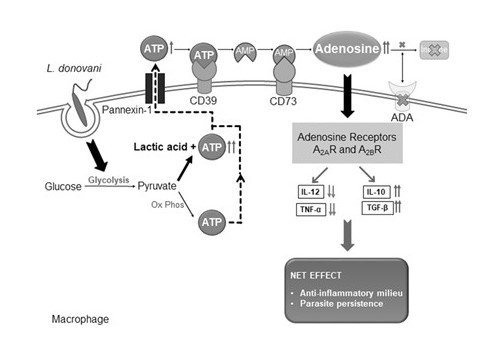
Intracellular survival of Leishmania donovani demands rapid production of host ATP for its sustenance. However, a gradual decrease in intracellular ATP in spite of increased glycolysis suggests ATP efflux during infection. Accordingly, upon infection, we show here that ATP is exported and the major exporter was pannexin-1, leading to raised extracellular ATP levels. Extracellular ATP shows a gradual decrease after the initial increase, and analysis of cell surface ATP-degrading enzymes revealed induction of the ectonucleotidases CD39 and CD73. Ectonucleotidase-mediated ATP degradation leads to increased extracellular adenosine (eADO), and inhibition of CD39 and CD73 in infected cells decreased adenosine concentration and parasite survival, documenting the importance of adenosine in infection. Inhibiting adenosine uptake by cells did not affect parasite survival, suggesting that eADO exerts its effect through receptor-mediated signalling. We also show that Leishmania induces the expression of adenosine receptors A2AR and A2BR, both of which are important for anti-inflammatory responses. Treating infected BALB/c mice with CD39 and CD73 inhibitors resulted in decreased parasite burden and increased host-favourable cytokine production. Collectively, these observations indicate that infection-induced ATP is exported, and after conversion into adenosine, propagates infection via receptor-mediated signalling.
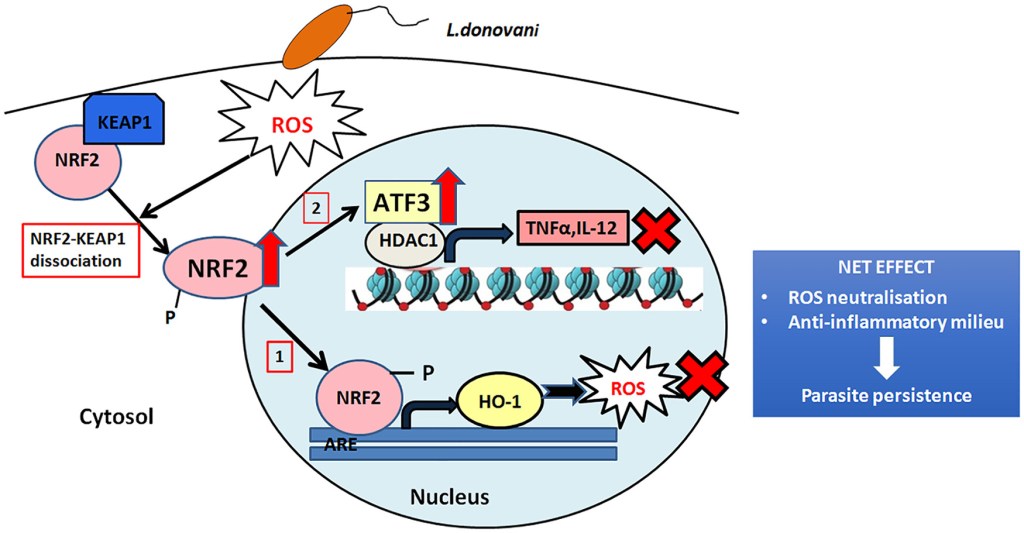
We showed previously that antioxidant enzyme heme oxygenase 1 (HO-1) is critical for Leishmania survival in visceral leishmaniasis. HO-1 inhibits host oxidative burst and inflammatory cytokine production, leading to parasite persistence. In the present study, screening of reported HO-1 transcription factors revealed that infection upregulated (4.1-fold compared to control [P < 0.001]) nuclear factor erythroid 2 (NFE2)-related factor 2 (NRF2). Silencing of NRF2 reduced both HO-1 expression and parasite survival. Investigation revealed that infection-induced transient reactive oxygen species (ROS) production dissociated NRF2 from its inhibitor KEAP1 and enabled phosphorylation-dependent nuclear translocation. Both NRF2 and HO-1 silencing in infection increased production of proinflammatory cytokines. But the level was greater in NRF2-silenced cells than in HO-1-silenced ones, suggesting the presence of other targets of NRF2. Another stress responsive transcription factor ATF3 is also induced (4.6-fold compared to control [P < 0.001]) by NRF2 during infection. Silencing of ATF3 reduced parasite survival (59.3% decrease compared to control [P < 0.001]) and increased proinflammatory cytokines. Infection-induced ATF3 recruited HDAC1 into the promoter sites of tumor necrosis factor alpha (TNF-α) and interleukin 12b (IL-12b) genes. Resulting deacetylated histones prevented NF-κB promoter binding, thereby reducing transcription of inflammatory cytokines. Administering the NRF2 inhibitor trigonelline hydrochloride to infected BALB/c mice resulted in reduced HO-1 and ATF3 expression, decreased spleen and liver parasite burdens, and increased proinflammatory cytokine levels. These results suggest that Leishmania upregulates NRF2 to activate both HO-1 and ATF3 for disease progression.
Funding Agency


